A new role for an old Alzheimer’s-related protein

Loss of memory is a well-established symptom of Alzheimer’s disease. Also, cognitive impairment and personality changes are also observed in these patients [1]. Many studies investigating the causes of Alzheimer’s have focused on the biochemical pathways and molecules involved in the pathology of the disease [2]. One of the main goals of such studies is to understand how amyloid plaques and neurofibrillary tangles, two of the biochemical hallmarks of Alzheimer’s disease pathology, lead to the brain degeneration typically observed in these patients. Plaques and tangles are abnormal aggregates of proteins and are believed to be toxic to nearby neurons, leading to the loss of both brain cells and their connections, which may play a major role in the onset and progression of Alzheimer’s. Another popular avenue of research focuses on changes at the microscopic scale that may relate to the loss of memory with the progression of the disease. Such studies have focused on the connections between neurons, anatomy of the brain regions involved, and molecules that may participate in the signaling between cells of the brain. Recent research published in PLOS ONE out of the University of Illinois at Chicago from the lab of Orly Lazarov shows that the Presenilin-1 protein, a protein linked to familial Alzheimer’s and to plaque formation, plays an important role in the birth of new neurons in the hippocampus, a brain structure crucial for memory formation and implicated in the disease [3]. Moreover, this research shows how loss of Presenilin-1 in newborn hippocampal cells can cause deficits in behavioral tests associated with memory.
Presenilin-1 affects on neurons and behavior
To determine the role of Presenilin-1 in the birth of new neurons in the hippocampus, the lead author of the study, Jacqueline Bonds, and her colleagues used a technique that allowed them to fluorescently tag neurons while also reducing levels of Presenilin-1 in a specific part of the hippocampus known as the dentate gyrus. In order to study the effects of reduced Presenilin-1, behavioral tasks were carried out at three months post-manipulation, a time point when many fluorescently labeled cells were observed. The first task measured whether an animal is able to distinguish between two similar contexts based on different cues provided by the researchers. For example, think of two rooms with similar layouts and dimensions but with different colored walls and with different odors. Such tasks are known to depend on the generation of new neurons in the hippocampus. At the three month mark, lower levels of Presenilin-1 did not seem to have much of an affect on the ability of the mice to successfully perform the task, as both regular mice and mice with reduced Presenilin-1 were able to distinguish between the two contexts successfully. However, at six months post-injection, the injected animals showed impairments in their ability to distinguish the separate contexts. This suggests that Presenilin-1 is important for the performance of this task.
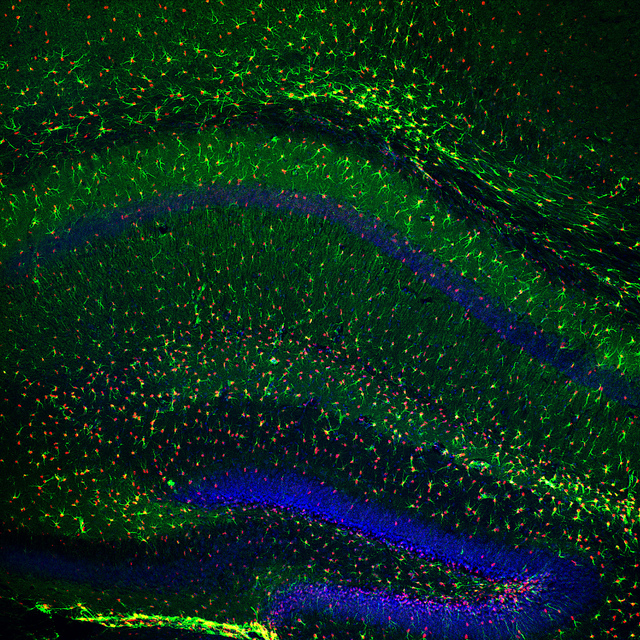
Since the scientists had shown that prolonged Presenilin-1 reduction affected a behavior that depends on the birth of new neurons in the hippocampus, they next wanted to test whether Presenilin-1 affects this process. Labeling cells with markers specific for newly generated neurons, Bonds et al. showed that there was a decrease in both mature neurons and newly generated mature neurons in the region of interest when Presenilin was reduced using the same method mentioned earlier. Similar decreases were observed in oligodendrocyte numbers, a supporting cell in the brain that was also examined. Next, the researchers took the study a step further and analyzed the shape of the surviving neurons to see whether or not this feature was also changed by the reduction of Presenilin-1. The authors noted significant decreases in the amount of branching in neurons as a result of reduced Presenilin-1 levels. Branches of these types of neurons are known to display tiny protrusions known as dendritic spines, which act as the site of contact and communication between two neurons. The authors also observed a decrease in the density of these dendritic spines. This decrease in spine density was observed anywhere from 10-20 microns from the cell soma. Mean length, surface area, and volume of spines were not significantly different when Presenilin was reduced. As mentioned, dendritic spines are important for communication between neurons, a process vital to normal brain function. Thus, the findings of this experiment suggest the ability of these neurons to signal to one another may be disrupted.
Evidence for the involvement of a signaling pathway
Up until this point, the paper had established clear affects of Presenilin-1 reduction on animal behavior (specifically behavior related to new hippocampal cells), cell number/survival in the hippocampus (as measured by the number of new neurons), as well as changes in dendritic branching and dendritic spine number. Such observations are important data in the understanding of Presenilin-1 involvement in neurogenesis and hippocampal-dependent memory function. However, such data provides little insight into the biochemical pathways linking Presenilin-1 to the changes observed in this study. Perhaps one of the most impressive pieces of this research is that Bond et al. were able to link Presenilin to a a specific pathway. To do this, the authors measured levels of molecules such as nestin, cyclin D1, EGFR, and neurofilament-L, and showed that they are changed in response to reduced Pesenilin-1. Specifically, neurofilament-L levels were significantly increased and nestin levels were significantly decreased. These molecules are involved in cell cycle regulation and thus the observed changes match previous experimental data generated during this study. The authors also mention they have previously observed changes in levels of Beta-catenin and notch-1 ligand delta when presenilin-1 is reduced [3]. In this study, using western blotting to quantify protein levels, the authors found that when presenilin-1 is reduced, levels of phospho-beta-catenin are changed, even though total levels of this protein are not different. Another protein, GSK3-beta was also probed as it is involved in the beta-catenin pathway; however, it was not altered. Finally, the authors also aimed to measure two additional role-players in the beta-catenin pathway, neither of which was convincingly altered, though seemingly consistent (but non-significant) trends in altered levels of these molecules were observed.
This study by Bond et al. provides a set of interesting results linking Presenilin-1 to important factors of hippocampal function at the molecular, cellular, network, and behavioral levels. Examining how such a protein can play a role at multiple levels earns this research group applause, as such a study is not always an easy task to complete. Though insights into the exact pathways mediating the cellular and behavioral deficits observed in the early experiments of this paper are incomplete, the authors have built a solid foundation for further investigating such pathways in the future. Convincing cellular phenotypes (i.e. reduced dendrite branching and reduced dendritic spines) as well as measurable behavioral outcomes in the behavioral task, provide Bond et al., as well as other researchers, with a set of deficits that can be used to measure how altering various molecules involved in relevant pathways either worsen or rescue this set of deficits. As Presenilin-1 is a known Alzheimer’s protein, future studies into these pathways may help lead to the identification of therapeutic targets for Alzheimer’s disease and other forms of dementia and neurodegeneration.
The big picture
Though overlooked for a long time, evidence for the birth of new neurons in the adult brain, especially in the hippocampus, has accumulated relatively recently. Though their exact role has not been fully worked out, it seems that these newborn cells may play a role in brain plasticity and learning and memory [4]. As Alzheimer’s disease involves progressive loss of memory and cell death in the hippocampus, the process of hippocampal neurogenesis has been a topic of interest for many Alzheimer’s researchers. Interestingly, various Alzheimer-associated proteins have been linked to this process, including the enzyme BACE and Presenilin-1 [5]. Earlier studies have shown that the loss of Presenilin-1 in mice can lead to impaired neurogenesis and disrupt the structural architecture of brain areas associated with newborn neurons [5]. In relation to these previous findings, the Bond study described above sheds some interesting light on the cellular and molecular changes that the loss of Presenilin-1 has on newborn neurons in the hippocampus. The deficits in neuron maturity and the finding of disrupted branching adds a new layer of insight into this growing body of literature.
Overall, Alzheimer’s represents a complex disease with some obvious genetic aspects (ie Presenilin-1) that may be harnessed to uncover common deficits or signaling pathways between genetic forms of Alzheimer’s and idiopathic forms. Moreover, the neurodegenerative effects of the disease make curbing, stopping, or even curing its progression a hard problem for the fields of medicine and science. Ideally, targeting cellular or chemical processes before the onset of degeneration may be helpful as the tissue may still be healthy and viable. This preventative approach is attractive, but before it can be realized, appropriate drug targets and disease processes must be pinned down. In this sense, studies like the ones discussed above provide a step forward in targeting useful cellular processes (such as neurogenesis) to cure disease. It will be interesting to see how other groups add to the set of data presented in the Bond paper and others to potentially take advantage of such processes.
References
Bonds, J. A., Kuttner-Hirshler, Y., Bartolotti, N., Tobin, M. K., Pizzi, M., Marr, R., & Lazarov, O. (2015). Presenilin-1 Dependent Neurogenesis Regulates Hippocampal Learning and Memory. PLoS ONE, 10(6), e0131266. http://doi.org/10.1371/journal.pone.0131266.s001
Lazarov, O., & Marr, R. A. (2010). Experimental Neurology. Experimental Neurology, 223(2), 267–281. http://doi.org/10.1016/j.expneurol.2009.08.009
Rodríguez, J. J., & Verkhratsky, A. (2011). Neurogenesis in Alzheimer’s disease. Journal of Anatomy, 219(1), 78–89. http://doi.org/10.1111/j.1469-7580.2011.01343.x
Sheng, M., Sabatini, B. L., & Sudhof, T. C. (2012). Synapses and Alzheimer’s Disease. Cold Spring Harbor Perspectives in Biology, 4(5), a005777–a005777. http://doi.org/10.1101/cshperspect.a005777
Small, D. H. (2008). Network dysfunction in Alzheimer’s disease: does synaptic scaling drive disease progression? Trends in Molecular Medicine, 14(3), 103–108. http://doi.org/10.1016/j.molmed.2007.12.006
The phosphatidylinositol 3-kinase/Akt pathway is likely a key player in neurogenesis in the hippocampus.
https://www.gcbi.com.cn/gclib/html/pubmed/detail/18616990
Presenilin 1 gene mutations inhibit the activation of this pathway.
http://www.jneurosci.org/content/19/13/5360.full
Peroxynitrites, the principal oxidant in Alzheimer’s disease also inhibits this pathway via nitration.
http://www.ncbi.nlm.nih.gov/pubmed/16410804
The inhibition of the phosphatidyinositol 3-kinase/Akt pathway also results in restricted blood flow in the brain, increased hyperphosphorylation and nitration of tau proteins which limits neurotransmissions and the transport of nutrients in the brain, and lowers levels of antioxidants in the brain.
Alzheimer’s disease can likely be treated with powerful peroxynitrite scavengers which also help to partially reverse oxidation and nitration. This includes eugenol in various essential oils (rosemary, clove, bay laurel, lemon balm etc.) and ferulic acid in panax ginseng.
http://onlinelibrary.wiley.com/doi/10.1111/j.1479-8301.2009.00299.x/full
http://www.ncbi.nlm.nih.gov/pubmed/22780999
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3659550/